
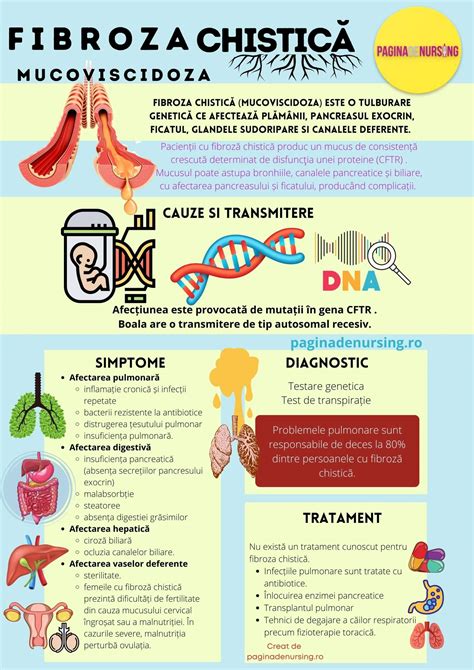
Fibroza chistică (FC) este o afecțiune genetică, transmisă ereditar, care afectează în principal sistemul respirator și cel digestiv. Cauzată de mutații în gena CFTR, boala determină producția de mucus gros și lipicios, care poate bloca pasajele din diverse organe, ducând la probleme de sănătate semnificative.
Fiecare persoană moștenește două copii ale genei CFTR, câte una de la fiecare părinte. Fibroza chistică apare atunci când ambele copii ale genei prezintă mutații. Părinții care au o singură copie a genei mutante sunt purtători sănătoși și nu suferă de afecțiune, dar pot transmite gena defectă copiilor lor.
Fibroza chistică este rezultatul unor mutații (modificări) în gena CFTR. Fiecare persoană are, în mod normal, două copii ale genei CFTR, o copie moștenită de la mamă și o copie moștenită de la tată. Fibroza chistică apare atunci când ambele copii ale genei CFTR prezintă mutații. Acest lucru înseamnă că părinții care prezintă, fiecare o mutație într-o singură copie a genei CFTR nu suferă de această afecțiune, însă pot avea împreună un copil cu fibroză chistică.
Deși fibroza chistică este progresivă și necesită gestionarea zilnică a simptomelor, persoanele afectate pot, de obicei, să aibă o viață aproape obișnuită, să meargă la școală și să lucreze. Calitatea vieții persoanelor afectate de fibroză chistică este mai bună în prezent decât cea a persoanelor diagnosticate câteva decenii în urmă.
Mecanismul de acțiune al bolii
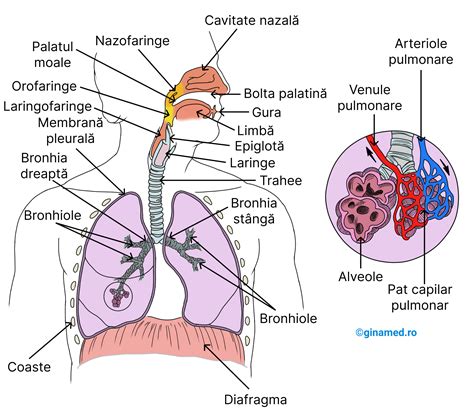
Sistemul anormal de transport al electroliților întâlnit în FC determină ca celulele din sistemul respirator, în special de la nivelul plămânilor, să absoarbă mai mult sodiu și apă decât au nevoie. FC manifestă la nivelul sistemului gastro-intestinal afectează în principal pancreasul, o glandă mixtă endocrină și exocrină. Secrețiile din pancreas devin, de asemenea, groase și pot obstrua canalele pancreasului. Acest lucru poate cauza o scădere a secreției de enzime la nivelul pancreasului. Ficatul poate fi, de asemenea, afectat.
Din cauza ratei ridicate de infecție în tractul respirator inferior, persoanele cu FC pot dezvolta o tuse cronică, sânge în spută și, adesea, chiar un plămân colapsat.
Majoritatea feților de gen masculin cu fibroză chistică au blocaje la nivelul canalului spermatic. Acest aspect patologic rezultă din secrețiile groase care blochează canalele deferente și le împiedică să se dezvolte corespunzător. Aceasta provoacă infertilitate la maturitate deoarece sperma nu poate parcurge traseul normal întâlnit în procesul de ejaculare. Există unele tehnici mai noi care permit bărbaților cu fibroză chistică să aibă copii.
Simptomele Fibrozei Chistice
Simptomele fibrozei chistice variază în funcție de severitatea bolii, care este determinată de mutațiile specifice din gena CFTR. Unii copii pot prezenta simptome încă de la naștere, în timp ce alții pot fi diagnosticați abia la vârsta adultă, manifestând simptome atipice precum pancreatită, infertilitate sau pneumonie recurentă.
Simptome respiratorii:
- Tuse persistentă care produce mucus gros (spută)
- Respirație șuierătoare
- Intoleranță la efort fizic
- Infecții pulmonare repetate
- Nas înfundat sau inflamat, sinuzite cronice
Simptome digestive:
- Scaune mirositoare și grele (steatoreice)
- Creștere lentă în greutate și înălțime
- Blocaj intestinal (în special la nou-născuți)
- Pancreatită
- Constipație severă, care poate duce la prolaps rectal
Persoanele cu fibroză chistică au un nivel de sare în transpirație mai mare decât cel normal.
Diagnosticarea Fibrozei Chistice
Diagnosticul fibrozei chistice implică o serie de teste, începând de la screening-ul nou-născuților și continuând cu teste specifice.
Testul sudorii (testul transpirației) este metoda standard de diagnosticare a fibrozei chistice și măsoară cantitatea de sare (concentrația de ioni de clor și sodiu) din transpirația individului. Există mai multe metode de colectare a transpirației, niciuna nu este dureroasă sau periculoasă. Dacă această cantitate de sare este anormal de mare, atunci este foarte probabil ca diagnosticul sa fie cel de fibroză chistică. Valorile normale ale electroliților în transpirație sunt cele care se situează sub valoarea de 40mMol/L; valorile pozitive la copii sunt peste 60 mMol/L, iar la adolescenți și adulți tineri peste 70 mMol/lL; în cazul valorilor echivoce, înscrise între 40-60 mMol/L testul se repetă obligatoriu și se interpreteză în context clinic. O concentrație de clorură de sodiu mai mare de 60 mMol/L în sudoare, la două determinări diferite, stabilește diagnosticul de boală.

Teste de screening la nou-născuți: IRT (trypsinogen imunoreactiv) - detectează nivelul unei enzime pancreatice. Dacă rezultatele sunt pozitive, se recomandă teste genetice.
Teste genetice: Sunt disponibile pentru testarea persoanelor asimptomatice care doresc să afle dacă sunt sau nu purtătoare ale genei defective de fibroză chistică. Aceste teste implică, de obicei, consiliere pre și post testare. Dacă investigațiile arată că o persoană este purtătoare a genei defective de fibroză chistică, este necesară și testarea partenerului.
Colegiul American de Genetică Medicală recomandă screening-ul purtătorilor utilizând un panel care pune în evidență 23 de mutații, în care sunt incluse majoritatea mutațiilor ce au o frecvență mai mare de 0.1% în populația generală din SUA.
Teste de diagnostic prenatal: Acestea pot identifica mutațiile genetice la un copil nenăscut, prin amniocenteză sau biopsie de vilozități coriale.
Înțelegerea fibrozei chistice (include fiziopatologia și diagnosticul)
Tratamentul Fibrozei Chistice
Deși nu există încă un tratament curativ pentru fibroza chistică, progresele înregistrate în ceea ce privește tratamentul și intervențiile asociate ajută oamenii să trăiască mai mult și mai sănătos. Managementul bolii este complex și necesită o abordare multidisciplinară, având ca obiective prevenirea și controlul infecțiilor pulmonare, eliminarea mucusului, tratarea blocajelor intestinale și asigurarea unei nutriții adecvate.
La nou-născuții cu un rezultat pozitiv la screening, tratamentul poate începe în timp ce diagnosticul este confirmat.
Metode de tratament:
- Tehnici de curățare a căilor respiratorii: Ajută la fluidizarea mucusului pulmonar pentru a facilita eliminarea acestuia, reducând infecțiile și îmbunătățind respirația. Acestea includ moduri speciale de respirație și tuse, dispozitive folosite pe gură și veste terapeutice care folosesc vibrații pentru a mobiliza mucusul, precum și terapie fizică toracică.
- Antibiotice: Previn sau tratează infecțiile pulmonare și îmbunătățesc funcția pulmonară.
- Medicamente antiinflamatorii: Reduc inflamația, care cauzează multe dintre modificările din fibroza chistică. Ibuprofenul este benefic în special pentru copii, dar efectele secundare pot induce probleme de creștere, renale și gastro-intestinale.
- Bronhodilatatoare: Relaxează și deschid căile respiratorii.
- Medicamente modulatoare ale CFTR: Influentează funcția proteinei CFTR defecte.
- Diluante de mucus: Facilitează eliminarea mucusului din căile respiratorii.
În 2019, FDA a aprobat primul medicament în combinație triplă (elexacaftor/ivacaftor/tezacaftor) pentru tratamentul pacienților cu cea mai frecventă mutație a genei CFTR, F508del, care afectează peste 90% dintre pacienți.
Deși nu există un tratament curativ pentru fibroza chistică, speranța de viață pentru persoanele afectate a crescut semnificativ datorită progreselor medicale, permițându-le să ducă o viață mai lungă și mai împlinită.
tags: #corion #cu #zone #de #fibroza